
ЙЩЦБМђГЦЃКЪцЬЉЩё ЙЩЦБДњТыЃК300204
ЙигкЪцЬЉЩёЃЈББОЉЃЉЩњЮяжЦвЉЙЩЗнгаЯоЙЋЫОЩъЧыЯђЬиЖЈЖдЯѓЗЂааЙЩЦБЕФЕкШ§ТжЩѓКЫЮЪбЏКЏЕФ
ЛиИДБЈИц
БЃМіЛњЙЙЃЈжїГаЯњЩЬЃЉ
ЃЈГЩЖМЪаЧрбђЧјЖЋГЧИљЩЯНж95КХЃЉ
ЖўЉЖўШ§ФъСљдТ
ЙигкЪцЬЉЩё(ББОЉ)ЩњЮяжЦвЉЙЩЗнгаЯоЙЋЫОЩъЧыЯђЬиЖЈЖдЯѓЗЂааЙЩЦБЕФЕкШ§ТжЩѓКЫЮЪбЏКЏЕФЛиИДБЈИц
ЩюлкжЄШЏНЛвзЫљЃК
ИљОнЙѓЫљгк2023Фъ6дТ1ШеГіОпЕФЁЖЙигкЪцЬЉЩёЃЈББОЉЃЉЩњЮяжЦвЉЙЩЗнгаЯоЙЋЫОЩъЧыЯђЬиЖЈЖдЯѓЗЂааЙЩЦБЕФЕкШ§ТжЩѓКЫЮЪбЏКЏЁЗЃЈЩѓКЫКЏЁВ2023ЁГ020088КХЃЉЃЌЪцЬЉЩёЃЈББОЉЃЉЩњЮяжЦвЉЙЩЗнгаЯоЙЋЫОЃЈвдЯТМђГЦЁАЪцЬЉЩёЁБЁЂЁАЙЋЫОЁБЁЂЁАЗЂааШЫЁБЛђЁАЩъЧыШЫЁБЃЉгыБЃМіЛњЙЙЙњН№жЄШЏЙЩЗнгаЯоЙЋЫОЃЈвдЯТМђГЦЁАБЃМіЛњЙЙЁБЃЉЁЂББОЉЪаПЕДяТЩЪІЪТЮёЫљЃЈвдЯТМђГЦЁАТЩЪІЁБЃЉЕШЯрЙиЗНЖдЩѓКЫЮЪбЏКЏЫљСаЮЪЬтНјааСЫж№ЯюКЫВщТфЪЕКЭЪщУцЫЕУїЃЌВЂЖдЪцЬЉЩёЃЈББОЉЃЉЩњЮяжЦвЉЙЩЗнгаЯоЙЋЫОЩъЧыЮФМўгаЙиФкШнНјааСЫБивЊЕФаоИФЁЂВЙГфЫЕУїЛђИќаТЃЌЯжЛиИДШчЯТЃЌЧыгшЩѓКЫЁЃ
ШчЮоЬиБ№ЫЕУїЃЌБОЛиИДБЈИцжаЕФМђГЦгыЁЖЪцЬЉЩёЃЈББОЉЃЉЩњЮяжЦвЉЙЩЗнгаЯоЙЋЫО2022ФъЖШЯђЬиЖЈЖдЯѓЗЂааЙЩЦБФММЏЫЕУїЪщЁЗжаЁАЪЭвхЁБЫљЖЈвхЕФМђГЦОпгаЯрЭЌКЌвхЃЌЫљгУзжЬхЖдгІФкШнШчЯТЃК
| ЩѓКЫЮЪбЏКЏЫљСаЮЪЬт | КкЬхЃЈМгДжЃЉ |
| ЩѓКЫЮЪбЏКЏЫљСаЮЪЬтЕФЛиИД | ЫЮЬхЃЈВЛМгДжЃЉ |
| ЖдФММЏЫЕУїЪщЕШЩъЧыЮФМўЕФаоИФЁЂВЙГф | ПЌЬхЃЈМгДжЃЉ |
| ЖдФММЏЫЕУїЪщЕШЩъБЈИхЮФМўЕФв§гУ | ЫЮЬхЃЈВЛМгДжЃЉ |
дкБОЛиИДжаЃЌШєКЯМЦЪ§гыИїЗжЯюЪ§жЕЯрМгжЎКЭдкЮВЪ§ЩЯДцдкВювьЃЌОљЮЊЫФЩсЮхШыЫљжТЁЃ
ФП ТМ
ФП ТМ ...... 2
ЮЪЬтвЛ ...... 3
ЦфЫћЮЪЬт ...... 11
ЮЪЬтвЛИљОнЩъБЈВФСЯЃЌБОДЮФМЭЖЯюФПSTSA-1002КЭSTSA-1005СЊКЯгУвЉЯюФПгк2022Фъ8дТШЁЕУСйДВЪдбщХњзМЭЈжЊЪщЃЌЪЪгІжЂЮЊжЮСЦжиаЭЁЂЮЃжиаЭаТаЭЙкзДВЁЖОЗЮбзЃЌИУЯюФПДІгкСйДВIЦкНзЖЮЁЃКѓајЃЌЗЂааШЫФтПЊеЙгыжЮСЦжиаЭЁЂЮЃжиаЭаТаЭЙкзДВЁЖОЗЮбзЗНЯђЮоЙиЕФМБадКєЮќОНЦШзлКЯжЂ(ARDSЪЪгІжЂ)II/IIIЦкСйДВЪдбщ,зюжеЪЪгІжЂЗНЯђЮЊARDSЁЃ
ЧыЗЂааШЫВЙГфЫЕУїЃКЗЂааШЫБфИќФМЭЖЯюФПВњЦЗЪЪгІжЂЗЖЮЇгыЯрЙивЉЮяСйДВХњМўФкШнВЛвЛжТЃЌЪЧЗёашвЊШЁЕУОГФкЭтЩѓХњВПУХЕФХњзМЛђБИАИЃЌЪЧЗёашвЊШЁЕУаТЕФСйДВХњМўЁЃШчЪЧЃЌЧыЗЂааШЫНсКЯгаЙиЙцЖЈЫЕУїЯрЙиГЬађЁЂЪЕЪЉНјеЙЁЂЭЌРрАИР§ЕШЫЕУїФМЭЖЯюФПЪЕЪЉЪЧЗёОпгажиДѓВЛШЗЖЈадЁЃЧыБЃМіЛњЙЙКЭЗЂааШЫТЩЪІКЫВщВЂЗЂБэУїШЗвтМћЁЃЛиИДЃК
вЛЁЂЪЧЗёашвЊШЁЕУОГФкЭтЩѓХњВПУХЕФХњзМЛђБИАИЃЌЪЧЗёашвЊШЁЕУаТЕФСйДВХњМў
БОДЮФМЭЖЯюФПSTSA-1002КЭSTSA-1005СЊКЯгУвЉЯюФПЃЈвдЯТМђГЦЁАСЊКЯгУвЉЯюФПЁБЃЉвбгк2022Фъ8дТШЁЕУЙњМввЉЦЗМрЖНЙмРэОжЃЈNMPAЃЉЧЉЗЂЕФСйДВЪдбщХњзМЭЈжЊЪщЃЌЪЪгІжЂЮЊжЮСЦжиаЭЁЂЮЃжиаЭаТаЭЙкзДВЁЖОЗЮбзЃЌКѓајФтЯђУРЙњЪГЦЗвЉЦЗМрЖНЙмРэОжЃЈFDAЃЉЩъЧыПЊеЙгыCOVID-19ЗНЯђЮоЙиЕФARDSЪЪгІжЂЕФСйДВбаОПЃЌЗЂааШЫЭиеЙARDSЪЪгІжЂашвЊШЁЕУFDAЕФаТСйДВХњМўЁЃ
1ЁЂЭиеЙЪЪгІжЂЕФПЩааад
вЛИіаТвЉИљОнЦфзїгУЛњРэЃЌПЩФмЪЪгУЖрИіФПБъЪЪгІжЂЁЃаТвЉбаЗЂЙЋЫОдкШЁЕУЪзИіЪЪгІжЂЕФСйДВХњМўКѓЃЌдкПЊеЙIЦкСйДВНЁПЕШЫШКЕФЪдбщЦкМфЃЌПЩИљОнВЛЭЌФПБъЪЪгІжЂЕФбаЗЂжмЦкЁЂбаЗЂФбЖШЁЂОКељзДПіЁЂПЭЙлЯожЦЕШЖржжвђЫиЃЌбЁдёгХЯШбаЗЂаТЕФЪЪгІжЂЁЃФПЧАСЊКЯгУвЉЯюФПДІгкСйДВIЦкНзЖЮЃЌ IЦкСйДВЙЄзїФкШнжївЊЮЊГѕВНЕФСйДВвЉРэбЇМАШЫЬхАВШЋадЦРМлЪдбщЃЌМДжївЊЙлВьвЉЦЗЕФАВШЋадЖјЗЧгааЇадЃЌвђДЫЪмЪдепЭЈГЃЮЊНЁПЕШЫШКЃЌЪЕМЪЩЯВЂВЛЧјЗжЪЪгІжЂЁЃЕБаТвЉбаОПЕФЪмЪдепЮЊФПБъЪЪгІжЂЛМепЪБЃЌаТвЉбаЗЂЙЋЫОВХЛсзюжеШЗЖЈЪЪгІжЂЃЌВЂвдЭиеЙЪЪгІжЂЕФЗНЪНШЁ
ЕУаТЪЪгІжЂЕФСйДВХњМўЁЃ
аТвЉбаЗЂЙЋЫОдкЭиеЙаТЪЪгІжЂЪБЃЌПЩвдЪЙгУдЪЪгІжЂIЦкСйДВЕФНЁПЕШЫШКЪдбщЪ§ОнЃЌЯђвЉМрВПУХжБНгЩъЧыПЊеЙеыЖдаТЪЪгІжЂЛМепЕФСйДВЪдбщЁЃвђДЫЃЌЭиеЙаТЪЪгІжЂЯЕЪЙгУСЫдЪЪгІжЂЕФЪдбщЪ§ОнЃЌЭиеЙЪЪгІжЂЕФСйДВЪдбщЗНАИЩшМЦгыЪзДЮЩъЧыЕФСйДВЪдбщЗНАИДцдкЧјБ№ЃЌдЪЪгІжЂЕФIЦкСйДВНЁПЕШЫШКЪдбщЪ§ОнЖдЭиеЙаТЪЪгІжЂОпгаживЊжЇГХзїгУЁЃ
2ЁЂЛёЕУNMPAСйДВЪдбщаэПЩКѓЃЌКѓајдкУРЙњжБНгПЊеЙФПБъЪЪгІжЂЛМепСйДВЪдбщЕФПЩааад
NMPAКЭFDAЯжОљЮЊICHЃЈЙњМЪШЫгУвЉЦЗзЂВсММЪѕвЊЧѓаЕїЛсЃЉЕФГЩдБЁЃICHЪЧвЛИіЙњМЪадЗЧгЏРћзщжЏЃЌвбГЩЮЊвЉЦЗзЂВсСьгђживЊЕФЙњМЪЙцдђжЦЖЈепЃЌЦфжМдкаЕїИїЙњЕФвЉЮязЂВсММЪѕвЊЧѓЃЈАќРЈЭГвЛБъзМЁЂМьВтвЊЧѓЁЂЪ§ОнЪеМЏМАБЈИцИёЪНЃЉЃЌЪЙвЉЮяЩњВњГЇМвФмЙЛгІгУЭГвЛЕФзЂВсзЪСЯЙцЗЖЃЌАДееICHЕФгааЇадЁЂжЪСПЁЂАВШЋадМАзлКЯбЇПЦжИФЯЩъБЈЁЃЦфжаЃЌICHЕФживЊШЮЮёАќРЈаЕїИїЙњМрЙмЛњЙЙЃЌЪЙбаЗЂЪ§ОнИќДѓГЬЖШЕФЯрЛЅНгЪмЃЌБмУтдкШЫЬхЩЯжиИДПЊеЙСйДВЪдбщЁЃ
ICHдквНвЉбаЗЂЕФжЪСПЁЂАВШЋадЁЂгааЇадМАЖрбЇПЦЕШСьгђжЦЖЉСЫжюЖрЕФжИЕМддђЃЌЦфжаАќРЈСЫE6ЃЈR1ЃЉвЉЮяСйДВЪдбщЙмРэЙцЗЖжИЕМддђЃЈGCPЃЉЁЂE6ЃЈR2ЃЉвЉЮяСйДВЪдбщЙмРэЙцЗЖзлКЯИНТМЁЂE5ЃЈR1ЃЉНгЪмЙњЭтСйДВЪдбщЪ§ОнЕФжжзхвђЫиЁЂE17ЖрЧјгђСйДВЪдбщМЦЛЎгыЩшМЦЕФвЛАуддђЕШЩцМАСйДВЪдбщБъзММАЙњМЪЛЅШЯЕФЯрЙижИЕМддђЁЃОпЬхФкШнШчЯТБэЫљЪОЃК
| жИЕМддђУћГЦ | жївЊФкШн | жИЕМддђЫљДІНзЖЮ |
| E6ЃЈR1ЃЉвЉЮяСйДВЪдбщЙмРэЙцЗЖжИЕМддђЃЈGCPЃЉЁЂE6ЃЈR2ЃЉвЉЮяСйДВЪдбщЙмРэЙцЗЖзлКЯИНТМ | 1ЁЂвЉЮяСйДВЪдбщжЪСПЙмРэЙцЗЖЃЈGCPЃЉЪЧЩшМЦЁЂЪЕЪЉЁЂМЧТМКЭБЈИцЩцМАШЫРрЖдЯѓВЮМгЕФЪдбщЕФЙњМЪадТзРэКЭПЦбЇжЪСПБъзМЁЃзёбетвЛБъзМЮЊБЃЛЄЪмЪдепЕФШЈРћЁЂАВШЋадКЭНЁПЕЃЌЮЊгыдДгкКеЖћаСЛљаћбдЕФддђБЃГжвЛжТвдМАСйДВЪдбщЪ§ОнЕФПЩаХадЬсЙЉСЫЙЋжкБЃжЄЁЃ 2ЁЂGCPжМдкЬсГівЛИіЭГвЛЕФСйДВЪдбщБъзМЃЌДйНјЙмРэЕБОждкЦфШЈЯоФкЯрЛЅНгЪмСйДВЪ§ОнЁЃ 3ЁЂдкгавтЯђЬсНЛИјвЉеўЙмРэЕБОжСйДВЪ§ОнЪБгІЕБзёбБОжИЕМддђЁЃ | Ек5НзЖЮзЂ |
| E5ЃЈR1ЃЉНгЪмЙњЭтСйДВЪдбщЪ§ОнЕФжжзхвђЫи | 1ЁЂБОжИФЯЕФЧАЬсЪЧЃЌУЛгаБивЊдкаТЕиЧјжиИДНјаавЉЮяЕФШЋВПСйДВбаЗЂЙ§ГЬЃЌНЈвщШЋВПЛђВПЗжЕФНгЪмЙњЭтСй | Ек5НзЖЮ |
| ДВбаОПЪ§ОнЃЌвджЇГжвЉЮядкаТЕиЧјЕФзЂВсЩѓХњЁЃ 2ЁЂБОжИФЯВЂВЛЪЧЮЊСЫдкаТЕиЧјЩъЧызЂВсвЉЦЗЖјаоИФЖдСйДВЪ§ОнЕФвЊЧѓЃЌЖјЪЧжМдкЙњЭтСйДВбаОПЪ§ОнПЩФмЗћКЯаТЕиЧјЕФзЂВсвЊЧѓЪБЃЌЭЦМіНгЪмЙњЭтСйДВзЪСЯЁЃ 3ЁЂШєЯжгаСйДВЪ§ОнМЏЗћКЯаТЕиЧјЕФЙмРэвЊЧѓЃЌетаЉЙњЭтЪ§ОнЪЧЗёФмБЛНгЪмЃЌЛЙШЁОігкИУЪ§ОнФмЗёЭтЭЦЕНаТЕиЧјЕФШЫШКЁЃЕБМрЙмЛњЙЙЛђЩъАьепШЯЮЊжжзхвђЫиПЩФмИФБфвЉЮядкаТЕиЧјШЫШКжаЕФАВШЋадЛђгааЇадЪБЃЌЩъАьепПЩФмашвЊдкаТЕиЧјЛёЕУвЛЖЈЕФСйДВЪ§ОнЃЌвдБуНЋСНИіЕиЧјжЎМфЕФСйДВЪ§ОнЭтЭЦЛђЧХНгЦ№РДЁЃ 4ЁЂШчЙћЩъАьепашвЊЛёЕУЖюЭтЕФСйДВЪ§ОнЃЌвдТњзуаТЕиЧјЕФМрЙмвЊЧѓЃЌдђПЩвдНЋетаЉСйДВЪдбщЩшМЦГЩЧХНгбаОПЃЈbridging studyЃЌаЁЙцФЃЕФИНМгЪдбщбаОПЃЉЁЃ | ||
| E17ЖрЧјгђСйДВЪдбщМЦЛЎгыЩшМЦЕФвЛАуддђЕШЩцМАСйДВЪдбщБъзМ | 1ЁЂЫцзХвЉЮябаЗЂШевцШЋЧђЛЏЃЌВЛЭЌЧјгђКЭЙњМвМрЙмЛњЙЙФмЙЛНгЪмЖрЧјгђСйДВЪдбщЃЈMulti-Regional Clinical TrialsЃЌMRCTЃЉЪ§ОнзїЮЊжЇГжвЉЮяЃЈвНвЉВњЦЗЃЉХњзМЩЯЪаЕФжївЊжЄОнРДдДвбОБфЕУКмживЊЁЃБОжИЕМддђжМдкЫЕУїMRCTМЦЛЎгыЩшМЦЕФвЛАуддђЃЌФПЕФЪЧЬсИпMRCTдкШЋЧђМрЙмЕнНЛжаЕФПЩНгЪмЖШЁЃ 2ЁЂБОжИЕМддђжївЊеыЖдMRCTЃЌЦфЪ§ОнгУгкЬсНЛжСЖрИіМрЙмЛњЙЙЕФвЉЮяХњзМЃЈАќРЈХњзМдіМгЪЪгІжЂЁЂаТМСаЭвдМАаТИјвЉЗНАИЃЉЛђТњзуЩЯЪаКѓвЊЧѓЕФбаОПЁЃБОжИЕМддђЕФВПЗжФкШнвВПЩФмгыСйДВдчЦкбаЗЂЛђКѓЦкбаОПЯрЙиЁЃБОжИЕМддђжївЊКИЧАќРЈЩњЮяжЦЦЗдкФкЕФвЉЮяЁЃ 3ЁЂВЮгыMRCTЕФЫљгабаОПжааФгІТњзуЪЪгУЕФжЪСПЁЂТзРэМАМрЙмБъзМЁЃгШЦфЪЧЃЌMRCTгІдкЫљгаЧјгђКЭбаОПжааФАДееICH GCPБъзМЪЕЪЉЃЌАќРЈЪдбщжааФгІдЪаэМрЙмЛњЙЙЪЕЪЉGCPКЫВщЁЃ 4ЁЂЙФРјMRCTЩъАьепЭЌЯрЙиМрЙмЛњЙЙейПЊПЦбЇзЩбЏЛсЁЃЩЯЪіЛЅЖЏгІдкMRCTЕФМЦЛЎНзЖЮНјааЃЌЬжТлећЬхбаЗЂМЦЛЎЕФМрЙмвЊЧѓвдМАMRCTЪ§ОнгУгкжЇГжЩЯЪааэПЩЕФПЩНгЪмадЁЃ | Ек5НзЖЮ |
зЂЃКЕк5НзЖЮЮЊЪЕЪЉжИЕМддђЃЌМДICHИїЕиЧј/ЙњМвМрЙмЛњЙЙЭЈЙ§ИїздааеўГЬађЪЕЪЉжИЕМддђЁЃ
вРОнFDAЯрЙиЗЈЙцМАжИФЯ, ШчЙћТњзувдЯТЬѕМўЃЌFDA НЋНгЪмЮДдкУРЙњЬсНЛINDЕФОГЭтСйДВЪ§ОнЃКЃЈ1ЃЉбаОПзёбЁЖвЉЮяСйДВЪдбщжЪСПЙмРэЙцЗЖЁЗЃЈGCPЃЉ НјааЃЛЃЈ2ЃЉFDAШЯЮЊгаБивЊЃЌЛсЭЈЙ§ЯжГЁКЫВщРДбщжЄбаОПЪ§ОнЃЛЃЈ3ЃЉЦфЫћашвЊПМТЧЕФвђЫиЃКШчбаОПШЫШКЁЂСйДВЪЕМљЁЂЗЈЙцвЊЧѓЕШЗНУцЕФВювьЁЃзлЩЯЃЌдкICHжИЕМддђКЭFDAЯрЙиЗЈЙцМАжИФЯЕФПђМмЯТЃЌжаУРМрЙмЛњЙЙОљЮЊICHГЩдБЃЌжаЙњОГФкзёбGCPЩшМЦЕФСйДВЪдбщЃЌЦфЪдбщЪ§ОндкЦфЫћICHГЩдБ
ЫљЪєЙњЃЈШчУРЙњЃЉЃЌОпБИЕУЕНМрЙмЛњЙЙШЯПЩЕФЛљДЁЁЃСЊКЯгУвЉЯюФПЯЕзёбGCPЩшМЦЕФСйДВЪдбщЃЌЗЂааШЫПЩвдНсКЯСЊКЯгУвЉОГФкIЦкСйДВНЁПЕШЫШКЕФЪдбщЪ§ОнЃЌЯђFDAЩъЧыаТЪЪгІжЂСйДВХњМўЃЌжБНгПЊеЙеыЖдаТЪЪгІжЂЧвЪмЪдепЮЊВЁЛМепЕФСйДВЪдбщЁЃЗЂааШЫЕФЦфЫћбаЗЂЯюФПжаЃЌвВДцдкIЦкСйДВдкОГФкПЊеЙЃЌIIЦкСйДВжБНгдкОГЭтПЊеЙЕФЧщаЮЁЃШчBDB-001ЯюФПЃЌЦфжиаЭCOVID-19ЪЪгІжЂЕФIЦкСйДВЪдбщЃЈНЁПЕШЫШКЃЉдкОГФкЪЕЪЉЃЌКѓајдкЮїАрбРЁЂгЁЖШЕШВЮгыICHЕФЙњМвБЛдЪаэжБНгПЊеЙIIЦк/IIIЦкСйДВЪдбщЁЃ
3ЁЂСЊКЯгУвЉЯюФПЭиеЙЪЪгІжЂЪЕЪЉЕФПЩааад
МБадКєЮќОНЦШзлКЯеїЃЈARDSЃЉЪЧвЛжждкЖЬЪБМфФкЃЈ1жмФкЃЉЗЂЩњЕФМБадЁЂУжТўадЕФбзжЂадЗЮЫ№ЩЫЃЌгЩбЯжиИаШОЁЂДДЩЫЁЂанПЫЕШИїжжЗЮФкЭтжТВЁвђЫиЕМжТЃЌСйДВБэЯжЮЊКєЮќОНЦШЁЂЭчЙЬадЕЭбѕбЊжЂКЭКєЮќЫЅНпЃЌЮЊГЃМћЕФЮЃМАШЫРрНЁПЕЕФКєЮќЯЕЭГжижЂБэЯжжЎвЛЁЃЖржжЮЃЯевђЫиПЩгеЗЂARDSЃЌАќРЈЂйжБНгЗЮЫ№ЩЫвђЫиЃКбЯжиЗЮВПИаШОЃЌЮИФкШнЮяЮќШыЃЌЗЮДьЩЫЃЌЮќШыгаЖОЦјЬхЃЌбЭФчЁЂбѕжаЖОЕШЃЛЂкМфНгЗЮЫ№ЩЫвђЫиЃКбЯжиИаШОЃЌбЯжиЕФЗЧаиВПДДЩЫЃЌжижЂМБадвШЯйбзЃЌДѓСПзЊЪфбЊЃЌЬхЭтбЛЗЃЌУжЩЂадбЊЙмФкФ§бЊЕШЁЃ
STSA-1002ЪЧвЛжжжизщПЙШЫВЙЬхЕААзC5aЕФШЋШЫдДIgG1ЕЅПЫТЁПЙЬхЃЌвжжЦC5aПЩгааЇзшЖЯЦфЖджаадСЃЯИАћЕФЧїЛЏЁЂNETsаЮГЩЁЂЯЫЮЌЕААзЩњГЩЁЃзшЖЯжаадСЃЯИАћНщЕМЕФУтвпМЄЛюЁЂЖдбЊЙмФкЦЄКЭЩЯЦЄЯИАћЕФЦЦЛЕЁЂбЊЙмЭЈЭИаддіМгКЭФ§бЊЙ§ГЬЃЌПЩзшЖЯARDSЗЂЩњЕФЩЯгЮЛњжЦЃЌМѕЧсARDSЕФбзжЂЗДгІЁЃSTSA-1005ЪЧвЛжжШЋШЫдДIgG4ЕЅПЫТЁПЙЬхЃЌПЩвдгыGM-CSFRІСЬивьадНсКЯЃЌзшЖЯGM-CSFRІСМАЦфХфЬхGM-CSFЕФЯрЛЅзїгУЃЌДгЖјМѕЩйЙ§ЖШбзжЂКЭЭЈЙ§НЕЕЭВњЩњGM-CSFЕФЯИАћвђзгЫЎЦНВЂвжжЦЖргрЕФЫшЯЕЯИАћЕФДцЛюЁЃSTSA-1002ПьЫйМѕЩйЬьШЛУтвпЯИАћв§Ц№ЕФЙ§ЖШбзжЂЗДгІЃЌSTSA-1005ИЈжњМѕЩйЙ§ЖШбзжЂЗДгІЃЌВЂМѕЩйЩЯгЮЬьШЛУтвпЯИАћЕФЗжЛЏМАЙЧЫшЖЏдБЃЌСНепСЊКЯОпгааЭЌЧБСІЃЌЮЊСНвЉСЊКЯжЮСЦARDSЕФСйДВбаОПЬсЙЉСЫРэТлвРОнЁЃ
БОДЮСЊКЯгУвЉЯюФПКѓајФтЭиеЙжСгыCOVID-19ЮоЙиЕФARDSЪЪгІжЂЃЌARDSЕФжТВЁЛњжЦгыжижЂCOVID-19ЯрЫЦЃЌжижЂCOVID-19ГЃМћЯИАћвђзгЗчБЉКЭСмАЭЯИ
АћМѕЩйЃЌЪЧЛМепЬьШЛУтвпЙ§ЖШМЄЛюЖјЪЪгІадУтвпМЄЛюВЛзуЕФЬхЯжЃЌБОжЪЩЯЮЊЫшЯЕЯИАћЙ§ЖШМЄЛюЃЌгыЦфЫћвђЫив§Ц№ЕФИпбзжЂбЧаЭARDSЪЧвЛжТЕФЁЃвђДЫЃЌБОДЮСЊКЯгУвЉЯюФПДгзїгУЛњРэЩЯПДЃЌЭиеЙжСARDSЪЪгІжЂОпБИПЩааадЁЃ
СЊКЯгУвЉЯюФПвбгк2022Фъ8дТЛёзМПЊеЙСйДВЪдбщЃЌФПЧАСйДВIЦкЪмЪдепвбЭъГЩЫцЗУВЂГізщЃЌУЛгаЗЂЩњбЯжиВЛСМЪТМўЃЌЯдЪОАВШЋадСМКУЁЃЭЌЪБЃЌЗЂааШЫЖдSTSA-1005КЭSTSA-1002ЕЅвЉЕФдРэеЦЮеНЯЮЊГфЗжЃЌSTSA-1002зЂЩфвКдкжаЙњЁЂУРЙњСНЕивВвбПЊеЙЕФЕЅвЉIЦкСйДВбаОПЃЌвдМАSTSA-1005зЂЩфвКдкУРЙњвбПЊеЙЕФЕЅвЉIЦкСйДВбаОПЃЌОљЯдЪОАВШЋадСМКУЁЃ
ЛљгкДЫЃЌЗЂааШЫСЊКЯгУвЉЯюФПдкгЕгаНЯЮЊГфЗжЕФвЉЮяАВШЋадЪ§ОнЕФЛљДЁЩЯЃЌЩъЧыARDSаТЪЪгІжЂСйДВбаОПЛёЕУFDAХњзМЕФПЩФмадНЯДѓЁЃМДБуFDAПЩФмЛљгкICHжИЕМддђЁЖE5ЃЈR1ЃЉНгЪмЙњЭтСйДВЪдбщЪ§ОнЕФжжзхвђЫиЁЗЃЌвЊЧѓЗЂааШЫВЙГфНјааЧХНгбаОПЃЈаЁЙцФЃЕФИНМгЪдбщбаОПЃЉЃЌдкЧАЪіСйДВЪдбщЪЕЪЉОбщЕФЛљДЁЩЯЃЌЧХНгЪЕбщврВЛДцдкНЯДѓЪЕЪЉФбЖШЛђепЪЕЪЉеЯАЁЃ
злЩЯЃЌЗЂааШЫдкICHжИЕМддђЯТПЊеЙЕФСЊКЯгУвЉЯюФПСйДВIЦкЪдбщЃЌЪдбщЯдЪОвЉЮяАВШЋадСМКУЃЌдЄМЦОГФкСйДВIЦкЪдбщЪ§ОнЛёЕУFDAШЯПЩЕФПЩФмадНЯДѓЃЌЮоашдйжиИДПЊеЙАВШЋадЯрЙиЕФдчЦкСйДВЪдбщЁЃЗЂааШЫЛљгкЯжгаЕФСйДВЪдбщЪ§ОнЃЌЯђFDAЬсНЛКѓајСйДВЪдбщЩъЧыВЂЛёЕУFDAЙигкARDSЗНЯђЕФСйДВХњМўЃЌВЛДцдкЪЕжЪадеЯАЃЌВЛДцдкжиДѓВЛШЗЖЈадЁЃ
ЖўЁЂЯрЙиГЬађМАЪЕЪЉНјеЙ
еыЖдСЊКЯгУвЉЯюФПЃЌЗЂааШЫМЦЛЎдкжаУРСНЕиОљПЊеЙЙигкСЊКЯгУвЉЯюФПдкARDSЪЪгІжЂЗНЯђЕФвЉЮябаЗЂЙЄзїЁЃПМТЧЕНSTSA-1002КЭSTSA-1005ЕЅвЉОљвбдкУРЙњПЊеЙСЫСйДВIЦкЪдбщЃЌгаНЯЮЊГфЗжЕФСйДВЪдбщЪ§ОнЃЌвђДЫЃЌСЊКЯгУвЉЯюФПЯШМЦЛЎдкУРЙњЯђFDAЩъЧыARDSЪЪгІжЂЗНЯђЕФбаОПЁЃЗЂааШЫвВНЋдкОГФкПЊеЙСЊКЯгУвЉЯюФПARDSЪЪгІжЂЗНЯђЕФбаОПЃЌФтЪЙгУздгазЪН№ЭЖШыЃЌВЛзїЮЊБОДЮФМЭЖЯюФПЁЃ
ЃЈвЛЃЉЯрЙиГЬађ
ЗЂааШЫЭъГЩСЊКЯгУвЉСйДВIЦкЪдбщЪмЪдепЕФЫцЗУКЭГізщКѓЃЌПЊеЙЪ§ОнЧхРэЙЄ
зїЃЌНјааЪ§ОнЕФЭГМЦЗжЮіЃЌВЂЭъГЩСйДВIЦкЪдбщЕФзмНсЙЄзїЁЃ
ЗЂааШЫИљОнСйДВЪ§ОнНсЙћМАFDAЕФЙЕЭЈЗДРЁЧщПіЃЌЯђFDAЕнНЛе§ЪНЕФINDЩъЧыВФСЯЁЃFDAНгЪмINDЩъЧыКѓЃЌЪЕЪЉЦРЩѓЁЃЭЈЙ§ЦРЩѓКѓЃЌFDAНЋХњзМЩъЧыШЫЕФСйДВЪдбщЗНАИЁЃ
ЃЈЖўЃЉЪЕЪЉНјеЙ
СЊКЯгУвЉЯюФПЕФСйДВIЦкЪдбще§дкЪЕЪЉжаЃЌНижСФПЧАЃЌвбШызщ50Р§НЁПЕШЫШКЪмЪдепЃЌОљвбЭъГЩЫцЗУВЂГізщЃЌУЛгаЗЂЩњбЯжиВЛСМЪТМўЃЌЯдЪОАВШЋадСМКУЃЌдЄМЦдк2023ФъЯТАыФъНјааСйДВIЦкЪдбщЕФзмНсЙЄзїЁЃ
ЗЂааШЫФПЧАдкгыFDAПЊеЙЛ§МЋЕФЙЕЭЈЃЌМЦЛЎдкЛёШЁСйДВIЦкЪдбщЪ§ОнКѓЃЌгк2023ФъЯТАыФъЯђFDAЕнНЛСЊКЯгУвЉаТЪЪгІжЂЕФINDЩъЧыЁЃ
Ш§ЁЂЭЌРраЭАИР§
аТвЉбаЗЂЦѓвЕПЊеЙаТвЉЕФдчЦкСйДВЪдбщЃЌКѓајЭиеЙаТЪЪгІжЂЃЌВЂБЛзМаэЛэУтдчЦкСйДВЃЌжБНгПЊеЙеыЖдаТЪЪгІжЂЛМепЕФСйДВЪдбщЃЌЯЕаТвЉбаЗЂЦѓвЕГЃгУЕФВйзїФЃЪНЁЃВПЗжЭЌРраЭАИР§ШчЯТБэЫљЪОЃК
| ЪЕЪЉжїЬх | вЉЦЗУћГЦ | дчЦкСйДВНзЖЮ | ЭиеЙЪЪгІжЂЕФСйДВНзЖЮ |
| вкЗЋвНвЉ ЃЈ002019ЃЉ | F-652 | СйДВНзЖЮЃКIЦкЃЛ ЪЕЪЉЕиЕуЃКОГФкЃЛ ЪЪгІжЂЃКМБадвШЯйбз | СйДВНзЖЮЃКIIЦкЃЌЛёЕУFDAЕФСйДВIIЦкЪдбщХњМўЃЌЛэУтIЦкЪдбщЃЛ ЪЕЪЉЕиЕуЃКУРЙњЃЛ ЪЪгІжЂЃКаТдіЪЪгІжЂМБадвЦжВЮяПЙЫожїВЁЁЂМБадОЦОЋИЮбз |
| ЩЯКЃвНвЉЃЈ601607ЃЉ | SPH4336 | СйДВНзЖЮЃКIЦкЃЛ ЪЕЪЉЕиЕуЃКОГФкЃЛ ЪЪгІжЂЃКЭэЦкЪЕЬхСі | СйДВНзЖЮЃКIIЦкЃЌЛёЕУFDAЕФСйДВIIЦкЪдбщХњМўЃЌЛэУтIЦкЪдбщЃЛ ЪЕЪЉЕиЕуЃКУРЙњЃЛ ЪЪгІжЂЃКОжВПЭэЦкЛђзЊвЦаджЌЗОШтСіЁЃ |
| ПЕКывЉвЕЃЈ002773ЃЉ | ПЕАиЮїЦе | СйДВНзЖЮЃКIЦк/IIЦкЃЛ ЪЕЪЉЕиЕуЃКОГФкЃЛ ЪЪгІжЂЃКжЮСЦЪЊадФъСфЯрЙиадЛЦАпБфад | СйДВНзЖЮЃКIIIЦкЃЌЛёЕУFDAЕФСйДВIIIЦкЪдбщХњМўЃЌЛэУтIЦкЁЂIIЦкЪдбщЃЛ ЪЕЪЉЕиЕуЃКУРЙњЃЛ ЪЪгІжЂЃКЪЊадФъСфЯрЙиадЛЦАпБфадЃЌКѓајЭиеЙжСаТЩњбЊЙмадФъСфЯрЙиадЛЦАпБфадЁЂЬЧФђВЁЛЦАпЫЎжзЃЈDMEЃЉЁЂЪгЭјФЄЗжжЇОВТізшШћЃЈBRVOЃЉЫљжТЛЦАпЫЎжзВЁБфЁЂЪгЭјФЄжабыОВТізшШћЃЈCRVOЃЉЫљжТЛЦАпЫЎжзВЁБфЕШЖрИіЪЪгІжЂ |
| ПЦТзвЉвЕ | A223 | СйДВНзЖЮЃКIЦк | СйДВНзЖЮЃКIIЦкЃЌЛёЕУNMPAЕФСйДВ |
| ЃЈ002422ЃЉ | ЪЕЪЉЕиЕуЃКОГФкЃЛ ЪЪгІжЂЃКРрЗчЪЊЙиНкбз | IIЦкЪдбщХњМўЃЌЛэУтIЦкЪдбщЃЛ ЪЕЪЉЕиЕуЃКОГФкЃЛ ЪЪгІжЂЃКжиЖШАпЭК | |
| жЧЯшН№ЬЉЃЈ688443ЃЉ | GR1501 | СйДВНзЖЮЃКIЦк ЪЕЪЉЕиЕуЃКОГФкЃЛ ЪЪгІжЂЃКАпПщзДвјаМВЁ | СйДВНзЖЮЃКIIЦкЃЈвбЭъГЩЃЉ/IIIЦкЃЈНјаажаЃЉЃЌЛёЕУNMPAЕФСйДВЪдбщХњМўЃЌЛэУтIЦкЪдбщЃЛ ЪЕЪЉЕиЕуЃКОГФкЃЛ ЪЪгІжЂЃКжажсаЭМЙжљЙиНкбз |
ДЫЭтЃЌСЊКЯгУвЉЯюФПЫфШЛЩаЮДШЁЕУARDSЕФСйДВХњМўЃЌЕЋвбШЁЕУCOVID-19ЯрЙиЪЪгІжЂЕФСйДВХњМўЃЌСйДВIЦкЕФбаЗЂЙЄзївбдкГжајНјаажаЃЌШЁЕУARDSЪЪгІжЂЕФаТСйДВХњМўВЂВЛЪЧИУЯюФПзїЮЊФМЭЖЯюФПЕФБиБИЬѕМўЁЃдкЪаГЁЭЌРрдйШкзЪЛђIPOАИР§жаЃЌВЛЩйвЉЦѓДцдкФМЭЖЯюФПЩаЮДШЁЕУСйДВХњМўЃЌЕЋвбЙцЛЎСйДВЭЖШыЕФЧщаЮЃЌШЁЕУСйДВХњМўВЂЗЧДДаТвЉбаЗЂРрФМЭЖЯюФПЕФБиБИЬѕМўЁЃВПЗжАИР§ОпЬхМћЯТБэЃК
| ЙЋЫОУћГЦ | ЗЂааЦЗжж | зЂВсЛђКЫзМШеЦк | ОпЬхЧщПі |
| РЅвЉМЏЭХЃЈSH.600422ЃЉ | ЙЋПЊЗЂааПЩзЊЛЛЙЋЫОеЎШЏ | 2020Фъ6дТ 28Ше | ФМЭЖЯюФПаТвЉбаЗЂЦНЬЈЩцМА6ИіаТВњЦЗЕФбаЗЂЃЌЦфжа4ИіВњЦЗKY41079ЁЂKY41111 ЁЂKY71113ЁЂKY70091ЩаЮДШЁЕУСйДВХњМўЃЌЕЋвбЙцЛЎСйДВЭЖШыЃЌВЂФтЪЙгУФММЏзЪН№ЁЃ |
| ОХЕфжЦвЉ ЃЈSZ.300705ЃЉ | ЙЋПЊЗЂааПЩзЊЛЛЙЋЫОеЎШЏ | 2021Фъ1дТ5Ше | ФМЭЖЯюФПаТвЉбаЗЂжаАќРЈPDX-02ЁЂPDX-03ЁЂPDX-04ЕШИФСМаЭаТвЉЃЌЩаЮДШЁЕУСйДВХњМўЃЌЕЋвбЙцЛЎСйДВЭЖШыЃЌВЂФтЪЙгУФММЏзЪН№ЁЃ |
| ШйВ§ЩњЮя ЃЈSH. 688331ЃЉ | ЪзДЮЙЋПЊЗЂааЙЩЦБ | 2022Фъ1дТ11Ше | ФМЭЖЯюФППЙжзСіПЙЬхаТвЉбаЗЂЯюФПЃЌЙВМЦ6ИівЉЮяЃЌЦфжаPD-L1+TGF-ІТЫЋПЙзЂЩфвКЃЌЩаЮДШЁЕУСйДВХњМўЃЌвбЙцЛЎСйДВIЦкжСIIIЦкЪдбщМАвЉЦЗзЂВсЗбгУЃЌВЂФтЪЙгУФММЏзЪН№ЁЃ |
| ПвђПЦММ ЃЈSH.688687ЃЉ | ЪзДЮЙЋПЊЗЂааЙЩЦБ | 2021Фъ1дТ5Ше | ФМЭЖЯюФПаТвЉбаЗЂжаЕФKW-027ЩаЮДШЁЕУСйДВХњМўЃЌЕЋвбЙцЛЎСйДВЧАбаОПЁЂСйДВIЦкжССйДВIIIЦкЕФЭЖШыЃЌВЂФтЪЙгУФММЏзЪН№ЁЃ |
злЩЯЃЌЗЂааШЫШЯЮЊЃК
ЃЈвЛЃЉБОДЮФМЭЖЯюФПSTSA-1002КЭSTSA-1005СЊКЯгУвЉЯюФПЃЌКѓајФтЯђFDAЩъЧыПЊеЙгыCOVID-19ЗНЯђЮоЙиЕФARDSЪЪгІжЂЕФСйДВбаОПЃЌашвЊШЁЕУFDAЕФаТСйДВХњМўЁЃЗЂааШЫдкICHжИЕМддђЯТПЊеЙЕФСЊКЯгУвЉЯюФПСйДВIЦкЪдбщЃЌФПЧАЪмЪдепвбЭъГЩЫцЗУВЂГізщЃЌЯдЪОвЉЮяАВШЋадСМКУЃЌдЄМЦОГФкСйДВIЦкЪдбщЪ§ОнЛёЕУICHГЩдБFDAШЯПЩЕФПЩФмадНЯДѓЃЌЮоашдйжиИДПЊеЙАВШЋадЯрЙиЕФдчЦкСйДВЪдбщЁЃЗЂааШЫЛљгкЯжгаЕФСйДВЪдбщЪ§ОнЃЌЯђFDAЬсНЛКѓајСйДВЪдбщЩъЧыВЂЛёЕУFDAЙигкаТЪЪгІжЂЕФСйДВХњМўЃЌВЛДцдкЪЕжЪадеЯАЁЃ
ЃЈЖўЃЉЯрНЯгкЦфЫћЯюФПЃЌСЊКЯгУвЉЯюФПОпгаЖРЬиЕФПЩаааджЇГжЛљДЁЁЃЦфвЛЃЌЪЪгІжЂДгCOVID-19ЭиеЙЕНARDSЃЌСЊКЯгУвЉЕФзїгУЭЈТЗКЭзїгУЛњжЦвРШЛгааЇЃЛЦфЖўЃЌSTSA-1002КЭSTSA-1005СНИіЕЅвЉЖМвбОЛёЕУFDAХњзМВЂПЊеЙСЫСйДВЪдбщЃЌВЂЧвIЦкСйДВОљБэЯжГіСМКУЕФАВШЋадЁЃ
ЃЈШ§ЃЉЛљгкЯжгаАИР§ЃЌаТвЉбаЗЂЦѓвЕПЊеЙаТвЉЕФдчЦкСйДВЪдбщЃЌКѓајЭиеЙаТЪЪгІжЂЃЌВЂБЛзМаэЛэУтдчЦкСйДВЃЌжБНгПЊеЙеыЖдаТЪЪгІжЂЛМепЕФСйДВЪдбщЃЌЯЕаТвЉбаЗЂЦѓвЕГЃгУЕФВйзїФЃЪНЁЃдкЪаГЁЭЌРрдйШкзЪЛђIPOАИР§жаЃЌВЛЩйвЉЦѓДцдкФМЭЖЯюФПЩаЮДШЁЕУСйДВХњМўЃЌЕЋвбЙцЛЎСйДВЭЖШыЕФЧщаЮЃЌШЁЕУСйДВХњМўВЂЗЧДДаТвЉбаЗЂРрФМЭЖЯюФПЕФБиБИЬѕМўЁЃвђДЫЃЌСЊКЯгУвЉЯюФПОЁЙмЮДШЁЕУARDSЪЪгІжЂЕФаТСйДВХњМўЃЌЕЋВЂВЛЛсвђДЫЯожЦИУЯюФПБЛСаШыФМЭЖЯюФПЁЃ
ЛљгкЩЯЪіЧщПіКЭЗжЮіЃЌБОДЮФМЭЖЯюФПSTSA-1002КЭSTSA-1005СЊКЯгУвЉЯюФПЯђFDAШЁЕУаТЪЪгІжЂЕФСйДВХњМўЃЌЪЕЪЉКѓајСйДВЪдбщЃЌВЛДцдкжиДѓВЛШЗЖЈадЁЃ
ЫФЁЂЗчЯеЬсЪО
ЗЂааШЫвбдкФММЏЫЕУїЪщЁАЕкЮхНк гыБОДЮЗЂааЯрЙиЕФЗчЯевђЫиЁБ/ЁАвЛЁЂЖдЙЋЫОКЫаФОКељСІЁЂОгЊЮШЖЈадМАЮДРДЗЂеЙПЩФмВњЩњжиДѓВЛРћгАЯьЕФвђЫиЁБ/ЁАЃЈЖўЃЉВњЦЗгыММЪѕЗчЯеЁБ/ЁА6ЁЂВњЦЗЙмЯпЯрЙиЗчЯеЁБжаЖдЯрЙиЗчЯегшвдВЙГфХћТЖЃЌОпЬхВЙГфХћТЖФкШнШчЯТЃК
ЁАЃЈ5ЃЉSTSA-1002КЭSTSA-1005СЊКЯгУвЉЯюФПЯрЙиЗчЯе
БОДЮФМЭЖЯюФПSTSA-1002КЭSTSA-1005СЊКЯгУвЉЯюФПвбгк2022Фъ8дТШЁЕУЙњМввЉЦЗМрЖНЙмРэОжЃЈNMPAЃЉЧЉЗЂЕФСйДВЪдбщХњзМЭЈжЊЪщЃЌЪЪгІжЂЮЊжЮСЦжиаЭЁЂЮЃжиаЭаТаЭЙкзДВЁЖОЗЮбзЃЌКѓајФтЯђУРЙњЪГЦЗвЉЦЗМрЖНЙмРэОжЃЈFDAЃЉЩъЧыПЊеЙгыCOVID-19ЗНЯђЮоЙиЕФARDSЪЪгІжЂЕФСйДВбаОПЃЌЗЂааШЫЭиеЙARDSЪЪгІжЂашвЊШЁЕУFDAЕФаТСйДВХњМўЁЃШєЗЂааШЫЮоЗЈЫГРћШЁЕУFDAЕФаТСйДВХњМўЃЌдђСЊКЯгУвЉЯюФПДцдкЮоЗЈНјвЛВНЭЦНјСйДВЪдбщЕФЗчЯеЁЃЁБ
ЮхЁЂжаНщЛњЙЙКЫВщГЬађМАКЫВщвтМћ
ЃЈвЛЃЉКЫВщГЬађ
БЃМіЛњЙЙМАЗЂааШЫТЩЪІжївЊжДааЕФКЫВщГЬађШчЯТЃК
1ЁЂЗУЬИЗЂааШЫИпМЖЙмРэШЫдБЁЂбаЗЂШЫдБЃЌСЫНтSTSA-1002КЭSTSA-1005СЊКЯгУвЉЯюФПЕФЪЕЪЉЧщПіМАКѓајашТФааЕФГЬађЃЛ
2ЁЂВщбЏICHЕФЛљБОЧщПіМАICHжЦЖЉЕФжИЕМадддђЃЛ
3ЁЂВщбЏFDAЯрЙиЗЈЙцЁЂжИФЯМАЙигкINDЩъЧыЕФЯрЙиеўВпЃЛ
4ЁЂВщбЏЭЌаавЕЙЋЫОЕФвЉЮябаЗЂЯюФПЃЌећРэгыСЊКЯгУвЉЯюФПЯрЫЦЕФАИР§ЁЃ
ЃЈЖўЃЉКЫВщНсТл
ОКЫВщЃЌБЃМіШЫМАЗЂааШЫТЩЪІШЯЮЊЃК
ЃЈвЛЃЉБОДЮФМЭЖЯюФПSTSA-1002КЭSTSA-1005СЊКЯгУвЉЯюФПЃЌКѓајФтЯђFDAЩъЧыПЊеЙгыCOVID-19ЗНЯђЮоЙиЕФARDSЪЪгІжЂЕФСйДВбаОПЃЌашвЊШЁЕУFDAЕФаТСйДВХњМўЁЃЗЂааШЫдкICHжИЕМддђЯТПЊеЙЕФСЊКЯгУвЉЯюФПСйДВIЦкЪдбщЃЌФПЧАЪмЪдепвбЭъГЩЫцЗУВЂГізщЃЌЯдЪОвЉЮяАВШЋадСМКУЃЌдЄМЦОГФкСйДВIЦкЪдбщЪ§ОнЛёЕУICHГЩдБFDAШЯПЩЕФПЩФмадНЯДѓЃЌЮоашдйжиИДПЊеЙАВШЋадЯрЙиЕФдчЦкСйДВЪдбщЁЃЗЂааШЫЛљгкЯжгаЕФСйДВЪдбщЪ§ОнЃЌЯђFDAЬсНЛКѓајСйДВЪдбщЩъЧыВЂЛёЕУFDAЙигкаТЪЪгІжЂЕФСйДВХњМўЃЌВЛДцдкЪЕжЪадеЯАЁЃ
ЃЈЖўЃЉЯрНЯгкЦфЫћЯюФПЃЌСЊКЯгУвЉЯюФПОпгаЖРЬиЕФПЩаааджЇГжЛљДЁЁЃЦфвЛЃЌЪЪгІжЂДгCOVID-19ЭиеЙЕНARDSЃЌСЊКЯгУвЉЕФзїгУЭЈТЗКЭзїгУЛњжЦвРШЛгааЇЃЛЦфЖўЃЌSTSA-1002КЭSTSA-1005СНИіЕЅвЉЖМвбОЛёЕУFDAХњзМВЂПЊеЙСЫСйДВЪдбщЃЌВЂЧвIЦкСйДВОљБэЯжГіСМКУЕФАВШЋадЁЃ
ЃЈШ§ЃЉЛљгкЯжгаАИР§ЃЌаТвЉбаЗЂЦѓвЕПЊеЙаТвЉЕФдчЦкСйДВЪдбщЃЌКѓајЭиеЙаТЪЪгІжЂЃЌВЂБЛзМаэЛэУтдчЦкСйДВЃЌжБНгПЊеЙеыЖдаТЪЪгІжЂЛМепЕФСйДВЪдбщЃЌЯЕаТвЉбаЗЂЦѓвЕГЃгУЕФВйзїФЃЪНЁЃдкЪаГЁЭЌРрдйШкзЪЛђIPOАИР§жаЃЌВЛЩйвЉЦѓДцдкФМЭЖЯюФПЩаЮДШЁЕУСйДВХњМўЃЌЕЋвбЙцЛЎСйДВЭЖШыЕФЧщаЮЃЌШЁЕУСйДВХњМўВЂЗЧДДаТвЉбаЗЂРрФМЭЖЯюФПЕФБиБИЬѕМўЁЃвђДЫЃЌСЊКЯгУвЉЯюФПОЁЙмЮДШЁЕУARDSЪЪгІжЂЕФаТСйДВХњМўЃЌЕЋВЂВЛЛсвђДЫЯожЦИУЯюФПБЛСаШыФМЭЖЯюФПЁЃ
ЛљгкЩЯЪіЧщПіКЭЗжЮіЃЌБОДЮФМЭЖЯюФПSTSA-1002КЭSTSA-1005СЊКЯгУвЉЯю
ФПЯђFDAШЁЕУаТЪЪгІжЂЕФСйДВХњМўЃЌЪЕЪЉКѓајСйДВЪдбщЃЌВЛДцдкжиДѓВЛШЗЖЈадЁЃЦфЫћЮЪЬт
вЛЁЂжиДѓгпЧщЧщПі
здЗЂааШЫБОДЮЗЂааЩъЧыгк2023Фъ3дТ6ШеЛёЩюНЛЫљЪмРэжСБОЛиИДГіОпШеЃЌЗЂааШЫГжајЙизЂУНЬхБЈЕРЃЌВЂЭЈЙ§ЭјТчМьЫїЕШЗНЪНЖдЙЋЫОБОДЮЗЂааЯрЙиУНЬхБЈЕРЧщПіНјааСЫздВщЃЌжївЊУНЬхБЈЕРМАЙизЂЪТЯюШчЯТЃК
| ађКХ | ШеЦк | УНЬхУћГЦ | ЮФеТБъЬт | жївЊЙизЂЪТЯю |
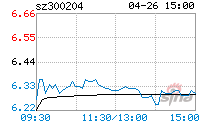
| 1 | 2023-03-07 | жаЙњЩЯЪаЙЋЫОЭј | ЪцЬЉЩёЖЈдіБЛЪмРэНЋгкЩюНЛЫљЩЯЪа | ЙЋЫОБОДЮЗЂааЩъЧывбЛёЩюНЛЫљЪмРэ |
| 2 | 2023-03-07 | жЄШЏжЎаЧ | ЪцЬЉЩёзюаТЙЋИцЃКЖЈдіЩъЧыЛёЩюНЛЫљЪмРэ | |
| 3 | 2023-03-16 | ИёТЁЛу | ЪцЬЉЩё(300204.SZ)ЃКОЭЩъЧыЖЈдіЁЂЪеЕНЩѓКЫЮЪбЏКЏ | ЩюНЛЫљЖдБОДЮЗЂааЕФЩъЧыЮФМўНјааЩѓКЫВЂЮЪбЏ |
| 4 | 2023-03-17 | НчУцаТЮХ | ЪцЬЉЩёЖЈдіВЛГЌ5.8вкдЊЪТЯюЪеЮЪбЏКЏЃКвЊЧѓЙЋЫОЫЕУїЪЧЗёЖдInflaRxДцдкжиДѓвРРЕ | ЪЧЗёЖдInflaRxДцдкжиДѓвРРЕ |
| 5 | 2023-03-20 | аТРЫВЦО | ЪцЬЉЩёЃК2022ФъОЛПїЫ№РЉжС1.37вкдЊЃЌЪцЬЉЧхЁЂЫеыФЩњЕШжївЊВњЦЗЪеШыЯТЛЌ | НтЖСЗЂааШЫ2022ФъФъЖШБЈИцЃЌжиЕуЙизЂОгЊвЕМЈЁЂбаЗЂЭЖШыЕШЧщПіЁЃ |
| 6 | 2023-03-20 | УПШеОМУаТЮХ | ЪцЬЉЩёЃК2022ФъЖШОЛРћШѓдМ-1.97вкдЊ | |
| 7 | 2023-03-20 | ЭкБДЭј | ЪцЬЉЩё2022ФъПїЫ№1.97вкЭЌБШПїЫ№діМг ЫеыФЩњЪЕЯжЯњЪлЪеШы | |
| 8 | 2023-03-21 | аТОЉБЈ | ЪцЬЉЩёЃК2022ФъЪеШы5.49вкдЊбаЗЂЭЖШыдьГЩЖЬЦквЕМЈГабЙ | |
| 9 | 2023-03-21 | жЄШЏШеБЈЭј | ЪцЬЉЩё2022ФъгЊЪеЪЕЯж5.49вкдЊНЋГжајЭЦНјдкбаЯюФПНјеЙ | |
| 10 | 2023-03-21 | аТРЫВЦО | гЅблдЄОЏЃКЪцЬЉЩёгЊвЕЪеШыЯТНЕ | |
| 11 | 2023-03-21 | ЭЌЛЈЫГВЦО | ВЦБЈЫйЕнЃКЪцЬЉЩё2022ФъШЋФъОЛПїЫ№1.97вкдЊЃЌзмЬхВЦЮёзДПіВЛМб | ЙизЂЗЂааШЫОгЊвЕМЈКЭВЦЮёзДПі |
| 12 | 2023-03-22 | вЉЖЩDaily | еѓЭДORаюСІ? | ЙизЂCOVID-19ЪЪгІжЂвЉЮябаЗЂЁЃ |
| 13 | 2023-03-22 | жаВЦЭј | ФъБЈ:ЪцЬЉЩёПїЫ№РЉДѓ ПлЗЧОЛРћШѓ-2.1вк | ЙизЂЗЂааШЫОгЊвЕМЈ |
| 14 | 2023-03-24 | юбУНЬх | баЗЂжЎТЗТўТўЃЌЛиБЈФбСЯ | ЙизЂЗЂааШЫРњДЮФМЭЖЯюФПБфЛЏЧщПігыCOVID-19ЪЪгІжЂвЉЮябаЗЂЁЃ |
| 15 | 2023-03-24 | аТРЫжЄШЏ | ЪцЬЉЩёвЕМЈГжајЖёЛЏЙщвђекекбкбкЃПЖЈдіФМЭЖЯюФПБЛМрЙмжЪвЩЮѓЕМЭЖзЪеп | ЪЧЗёЖдInflaRxДцдкжиДѓвРРЕЃЛФМЭЖЯюФПЪЧЗёЩцМАCOVID-19 |
| 16 | 2023-03-26 | юбУНЬх | Ш§ЦєЖЈдіЁЂСНЖШСїВњ | БОДЮФМЭЖЯюФПгыCOVID-19ЪЪгІжЂвЉЮябаЗЂЁЃ |
| 17 | 2023-04-06 | аТРЫВЦО | ВЮЙЩЙЋЫОInflaRxбаЗЂвЉЮягУгкжЮСЦCOVID-19ЛёЕУFDAНєМБЪЙгУЪкШЈ(EUA) | InflaRxбаЗЂвЉЮя VilobelimabгУгкжЮСЦCOVID-19 ЛёЕУFDA НєМБЪЙгУЪкШЈ |
| 18 | 2023-04-07 | Н№ШкНч | ЁАГйЕНЁБЕФаТЙквЉЭЈаажЄЮЊКЮФмИјЪцЬЉЩёДјРДаТЯыЯѓЃП | ПЯЖЈСЫC5aАаЕуЕФЮДРДЧАОА |
| 19 | 2023-04-25 | жЧЭЈВЦОЭј | ЪцЬЉЩёЃК2023ФъЕквЛМОЖШОЛРћШѓдМ-4873ЭђдЊ | ЙизЂвЛМОБЈвЕМЈЯТЛЌЧщаЮ |
| 20 | 2023-04-26 | НчУцаТЮХ | ЪцЬЉЩёЃКвЛМОЖШОЛПїЫ№ЭЌБШРЉДѓНќ1БЖЃЌЪцЬЉЧхЯњЪлЪеШыЯТНЕ |
еыЖдЩЯЪіЮЪЬтЃЌБЃМіЛњЙЙНјааСЫЯъЯИЕФКЫВщЃЌВЂОЭЯрЙиУНЬхжЪвЩЫљЩцЪТЯюНјааСЫНјвЛВНКЫВщЃЌОпЬхЧщПіШчЯТЃК
ЃЈвЛЃЉЙигкРњДЮдйШкзЪФМЭЖЯюФПБфЛЏЧщПі
ОпЬхЯъМћЗЂааШЫдк2023Фъ5дТ30ШегкОоГБзЪбЖЭјЃЈhttp://www.cninfo.com.cnЃЉЗЂВМЕФЁЖЙигкЪцЬЉЩёЃЈББОЉЃЉЩњЮяжЦвЉЙЩЗнгаЯоЙЋЫОЩъЧыЯђЬиЖЈЖдЯѓЗЂжЎааЙЩЦБЕФЩѓКЫЮЪбЏКЏЕФЛиИДБЈИцЃЈЖўДЮаоЖЉИхЃЉЃЈЛэУтАцЃЉЃЈ2023ФъвЛМОБЈВЦЮёЪ§ОнИќаТАцЃЉЁЗжЎЁАЮЪЬтвЛЁБжЎЁАЪЎЁБЁЃ
ЃЈЖўЃЉЙигкБЈИцЦкФквЕМЈЯТЛЌЧщПі
БЈИцЦкФкЗЂааШЫДІгкГжајПїЫ№зДЬЌЃЌжївЊЯЕЗЂааШЫГжајМгДѓбаЗЂЭЖШывдМАжївЊВњЦЗЯњЪлЪеШыЪмаавЕеўВпМАЪаГЁашЧѓгАЯьЫљжТЁЃОпЬхЯъМћЗЂааШЫдк2023Фъ5дТ30ШегкОоГБзЪбЖЭјЃЈhttp://www.cninfo.com.cnЃЉЗЂВМЕФЁЖЙигкЪцЬЉЩёЃЈББОЉЃЉЩњЮяжЦвЉЙЩЗнгаЯоЙЋЫОЩъЧыЯђЬиЖЈЖдЯѓЗЂжЎааЙЩЦБЕФЩѓКЫЮЪбЏКЏЕФЛиИДБЈИцЃЈЖў
ДЮаоЖЉИхЃЉЃЈЛэУтАцЃЉЃЈ2023ФъвЛМОБЈВЦЮёЪ§ОнИќаТАцЃЉЁЗжЎ ЁАЮЪЬтЖўЁБжЎЁАвЛЁБЁЃ
ЃЈШ§ЃЉЪЧЗёЖдInflaRxДцдкжиДѓвРРЕ
ЗЂааШЫЖдInflaRxВЛДцдкжиДѓвРРЕЃЌЫЋЗНЯЕЛљгквбЧЉЖЉЕФКЯзїавщЃЌПЊеЙЯрЙивЉЮяЕФбаЗЂЁЃОпЬхЯъМћЗЂааШЫдк2023Фъ5дТ30ШегкОоГБзЪбЖЭјЃЈhttp://www.cninfo.com.cnЃЉЗЂВМЕФЁЖЙигкЪцЬЉЩёЃЈББОЉЃЉЩњЮяжЦвЉЙЩЗнгаЯоЙЋЫОЩъЧыЯђЬиЖЈЖдЯѓЗЂжЎааЙЩЦБЕФЩѓКЫЮЪбЏКЏЕФЛиИДБЈИцЃЈЖўДЮаоЖЉИхЃЉЃЈЛэУтАцЃЉЃЈ2023ФъвЛМОБЈВЦЮёЪ§ОнИќаТАцЃЉЁЗжЎЁАЮЪЬтвЛЁБжЎЁАвЛЁБЁЃ
ЃЈЫФЃЉЙигкБОДЮФМЭЖЯюФПЪЧЗёЩцМАПЊеЙаТЙквЉЮябаЗЂ
БОДЮФММЏзЪН№ФтЪЕЪЉЕФЁАSTSA-1002КЭSTSA-1005СЊКЯгУвЉЯюФПЁБЃЌЯЕЛљгквбШЁЕУCOVID-19ЪЪгІжЂХњМўЯТПЊеЙЕФеыЖдНЁПЕШЫШКЕФСйДВIЦкЪдбщЪ§ОнЃЌКѓајФтПЊеЙARDSЪЪгІжЂЕФII/IIIЦкСйДВЪдбщЃЌзюжеЪЪгІжЂЗНЯђЮЊARDSЪЪгІжЂЁЃ
вђДЫЃЌSTSA-1002КЭSTSA-1005СЊКЯгУвЉЯюФПФтПЊеЙARDSЪЪгІжЂЗНЯђЕФвЉЮябаЗЂЃЌВЛЩцМАаТЙквЉЮябаЗЂЁЃ
ОпЬхЯъМћЗЂааШЫдк2023Фъ5дТ30ШегкОоГБзЪбЖЭјЃЈhttp://www.cninfo.com.cnЃЉЗЂВМЕФЁЖЙигкЪцЬЉЩёЃЈББОЉЃЉЩњЮяжЦвЉЙЩЗнгаЯоЙЋЫОЩъЧыЯђЬиЖЈЖдЯѓЗЂжЎааЙЩЦБЕФЩѓКЫЮЪбЏКЏЕФЛиИДБЈИцЃЈЖўДЮаоЖЉИхЃЉЃЈЛэУтАцЃЉЃЈ2023ФъвЛМОБЈВЦЮёЪ§ОнИќаТАцЃЉЁЗжЎЁАЮЪЬтвЛЁБжЎЁАЫФЁБЁЃ
ЃЈЮхЃЉЙигквЛМОЖШвЕМЈЯТЛЌЧщаЮ
ЗЂааШЫ2023ФъвЛМОЖШвЕМЈЯТЛЌЃЌжївЊЯЕКЫаФВњЦЗжЎвЛЕФЪцЬЉЧхЪмЕНМЏВЩеўВпЕФгАЯьЃЌЯњЪлЪеШыГіЯжЯТЛЌЁЃ
ЖўЁЂБЃМіЛњЙЙКЫВщвтМћ
еыЖдЧАЪіЪТЯюЃЌБЃМіЛњЙЙТФааЕФКЫВщГЬађШчЯТЃК
ЭЈЙ§ЭјТчМьЫїЕШЗНЪНМьЫїздЗЂааШЫБОДЮЗЂааЩъЧыЪмРэШежСБОЛиИДГіОпжЎШеЯрЙиУНЬхБЈЕРЕФЧщПіЃЌВщПДЪЧЗёгагыЗЂааШЫгаЙиЕФжиДѓгпЧщЃЌВЂгыБОДЮЗЂааЯрЙиЩъЧыЮФМўНјааЖдБШЁЃ
ОКЫВщЃЌБЃМіЛњЙЙШЯЮЊЃК
ЗЂааШЫБОДЮЗЂааЩъЧыЮФМўжагыУНЬхБЈЕРЙизЂЕФЮЪЬтЯрЙиЕФаХЯЂХћТЖецЪЕЁЂзМШЗЁЂЭъећЃЌВЛДцдкгІХћТЖЮДХћТЖЕФЪТЯюЁЃКѓајБЃМіШЫНЋГжајЙизЂгыЗЂааШЫБОДЮЗЂааЁЂЗЂааШЫздЩэЯрЙиЕФУНЬхБЈЕРЕШЧщПіЃЌШчГіЯжУНЬхЖдИУЯюФПаХЯЂХћТЖецЪЕадЁЂзМШЗадЁЂЭъећадЬсГіжЪвЩЕФЧщаЮЃЌБЃМіШЫНЋМАЪБНјааКЫВщВЂЖНДйЗЂааШЫзіЯргІДІРэЁЃ
ЃЈБОвГЮое§ЮФЃЌЮЊЪцЬЉЩёЃЈББОЉЃЉЩњЮяжЦвЉЙЩЗнгаЯоЙЋЫОЁЖЙигкЪцЬЉЩёЃЈББОЉЃЉЩњЮяжЦвЉЙЩЗнгаЯоЙЋЫОЩъЧыЯђЬиЖЈЖдЯѓЗЂааЙЩЦБЕФЕкШ§ТжЩѓКЫЮЪбЏКЏЕФЛиИДБЈИцЁЗжЎИЧеТвГЃЉ
ЪцЬЉЩёЃЈББОЉЃЉЩњЮяжЦвЉЙЩЗнгаЯоЙЋЫО
Фъ дТ Ше
ЃЈБОвГЮое§ЮФЃЌЮЊЙњН№жЄШЏЙЩЗнгаЯоЙЋЫОЁЖЙигкЪцЬЉЩёЃЈББОЉЃЉЩњЮяжЦвЉЙЩЗнгаЯоЙЋЫОЩъЧыЯђЬиЖЈЖдЯѓЗЂааЙЩЦБЕФЕкШ§ТжЩѓКЫЮЪбЏКЏЕФЛиИДБЈИцЁЗжЎЧЉеТвГЃЉ
БЃМіДњБэШЫЃК _____________ _____________
бЯ ЧП ТР Рз
ЙњН№жЄШЏЙЩЗнгаЯоЙЋЫО
Фъ дТ Ше
БЃМіЛњЙЙЖЪТГЄЩљУї
БОШЫвбШЯецдФЖСЪцЬЉЩёЃЈББОЉЃЉЩњЮяжЦвЉЙЩЗнгаЯоЙЋЫОБОДЮЩѓКЫЮЪбЏКЏЕФЛиИДЕФШЋВПФкШнЃЌСЫНтБОЛиИДЩцМАЮЪЬтЕФКЫВщЙ§ГЬЁЂБОБЃМіЛњЙЙЕФФкКЫКЭЗчЯеПижЦСїГЬЃЌШЗШЯБОБЃМіЛњЙЙАДееЧкУуОЁд№ддђТФааКЫВщГЬађ,ЩѓКЫЮЪбЏКЏЕФЛиИДВЛДцдкащМйМЧдиЁЂЮѓЕМадГТЪіЛђжиДѓвХТЉЃЌВЂЖдЩЯЪіЮФМўЕФецЪЕадЁЂзМШЗадЁЂЭъећадГаЕЃЯргІЕФЗЈТЩд№ШЮЁЃ
БЃМіЛњЙЙЖЪТГЄЃК _____________
ЃЈЗЈЖЈДњБэШЫЃЉ ШН дЦ
ЙњН№жЄШЏЙЩЗнгаЯоЙЋЫО
Фъ дТ Ше